Role of Topological Phases in Quantum Molecular Dynamics
Artur
Izmaylov
Assoc.
Professor,
Department
of
Chemistry
University
of
Toronto
Friday,
March
8,
2019
10:00
a.m.
C2-361 (Reading
Room)
Abstract:
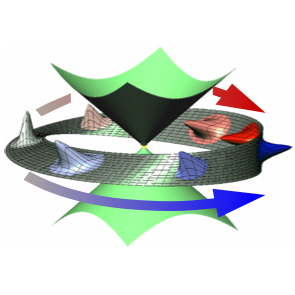
I start by considering the simplest model providing the CI topology: two-dimensional two-state linear vibronic coupling model. Selecting this model instead of a real molecule has the advantage that various dynamical regimes can be easily modeled in the model by varying parameters, whereas any fixed molecule provides the system specific behavior that may not be very illustrative. After demonstrating when topological phase effects are important and how they modify the dynamics for two sets of initial conditions (starting from the ground and excited electronic states), we give examples of molecular systems where the described topological phase effects are crucial for adequate description of nonadiabatic dynamics.
Understanding an extent of changes introduced by the topological phase in chemical dynamics poses a problem of capturing its effects by approximate methods of simulating nonadiabatic dynamics that can go beyond simple models. I assess the performance of both fully quantum (wave packet dynamics) and quantum-classical (surface-hopping, Ehrenfest, and quantum-classical Liouville equation) approaches in various cases where topological phase effects are important. It has been identified that the key to success in approximate methods is a method organization that prevents the quantum nuclear kinetic energy operator to act directly on adiabatic electronic wave functions.