Design team members: Adam Lau, Calvin Lau
Supervisor: Keith Ashman and Gary Bader
Background
In 1990, the U.S. Department of Energy and the National Institutes of Health began the U.S. Human Genome Project (HGP) - the identification and understanding of approximately 30,000 genes in the human DNA. The medical industry is building upon the knowledge, resources, and technologies emanating from this project to further understand the contributions of genetics to human health. The ultimate goal is to use this information to develop new ways to treat, identify, cure, or prevent diseases that affect humankind.
While mapping the human genome is a critical stage in the field of genomics, there is still much to be uncovered. At the direction of genes, human cells produce approximately 300,000 unique proteins. "The human genome isn't a blueprint but a building materials list. We know what components go into a cell, but what do they do? There is a host of interactions, and it's the interactions that make a cell healthy or unhealthy." explains biochemist Richard Caprioli (Hensley,S. 2001). The field of proteomics, a continuation of genomics, examines exactly this. Mass spectrometry is the primary tool used in proteomics research.
Project description
The area of proteomics research is still new and developing and a large number of manual and time consuming tasks exist which need to be addressed. Improvements in the area of systems integration of mass spectrometry and database searching will help streamline analysis procedures and greatly enhance the throughput of samples examined by researchers. This would be of overall interest to the field of proteomics. The project objectives are summarized below:
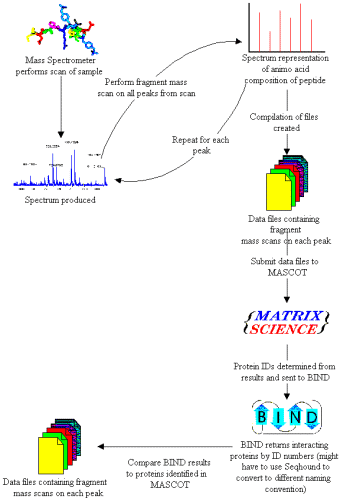
- mass spectrometer data passed to search engine
- protein identification results from search passed to BIND database to obtain known interaction partners
- proteins involved in any interactions found are examined for in the original results from the mass spectrometer
- any peptides (parts of a protein) not accounted for are further analyzed via mass spectrometry
- investigation of feasibility and performing searches over a LINUX cluster instead of a single unit
Design methodology
The majority of the activities involved with this project involve systems development - producing an information system solution to an organizational problem or opportunity (Laudon and Laudon, 2001). As shown in the diagram below, a six-stage cyclical systems development process will be used consisting of systems analysis, system design, programming, testing, conversion, and production as described by Laudon and Laudon.
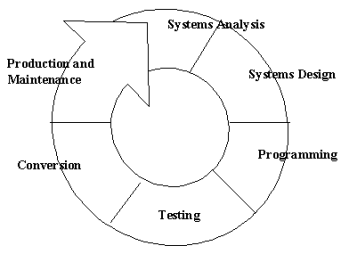
1. Systems analysis - Systems analysis involves the analysis and definition of the problem, identifying its causes, specifying the solution, and identifying the information requirements that must be met by a system solution.
2. Systems design - While systems analysis describes what a system should do to meet information requirements, systems design shows how the system will fulfill this objective. In this case, the proposed solution to the problem faced by SLRI may or may not change depending on the results of extensive research of the system's sub-components. Each sub-system (QA Analyst, MASCOT, and BIND) will be discussed in terms of compatibility and usability in the Evaluation section.
3. Programming - In the programming stage, system specifications prepared during the design stage are translated into software program code. The implementation stage is planned immediately after the evaluation of system sub-components. The proposed integration plan consists of four main systems: QA Analyst and the mass spectrometer, the MASCOT search database, the BIND protein interaction database, and the LINUX cluster (if implemented).
4. Testing - Exhaustive and thorough testing should be conducted on any new system implementation. This ensures that the system produces appropriate results for an acceptable range of inputs. In relation to this system, three types of testing will be performed - unit testing, system testing, and acceptance testing. The integration of the mass spectrometer, MASCOT, and BIND databases has never been performed and as a result, may be difficult to test without a base measurement. However, while such a system has never existed, the process of identifying protein interactions is currently ongoing at the institute. As a result, testing of the system will be compared to an already performed analysis of the yeast complex. This will allow accurate measurement of improvements in time and performance while maintaining the accuracy of results. Users of the system validate if the new system meets the specified requirements.
5. Conversion - Conversion is the process of changing from the old system to the new system. The implementation of the proposed solution requires no commissioning of current machinery and data. As a result, this stage is fairly minimal in terms of impact and resources.
6. Production and maintenance - This stage occurs when the new system is installed and conversion is complete. At this stage, the system is reviewed by both users and technical specialists to determine how well it has met its original objects and to decide whether any revisions or modifications are in order. Maintenance is then performed to make relevant changes in hardware, software, documentation, or procedures to correct errors, meet new requirements, or improve the overall system.
Related Links
-
Samuel Lunenfeld Research Institute Proteomics Lab
-
Matrix Science MASCOT Search Engine
-
MDS Sciex
-
BIND Database
-
Samuel Lunenfeld Research Institute