Background
To ensure sustained efficacy, novel therapeutics target conserved parts of the viral genome since these segments are less prone to viral mutations. Existing methods for detecting conserved parts of the viral genome are mainly based on sequence alignment that involve performing pairwise alignment between the predecessor and a descendant sequence and require extensive number of sequences that are rooted from the same ancestor and must be collected over time. Thus, when facing a novel virus or pathogen that has the potential to lead to a widespread epidemic or a global pandemic, this waiting process impedes the rapid development of targeted therapeutics that could have a critical impact on the case fatality rate or the magnitude of the outbreak. Importantly, the mutation information can seldom be extracted directly from the mutational changes observed in other members in the virus family. Due to these issues, there is a significant need to better understand how the pathogenesis of a virus is affected by viral mutations and to determine the conserved segments in the viral genome that can serve as stable targets for novel therapeutics.
Description of the invention
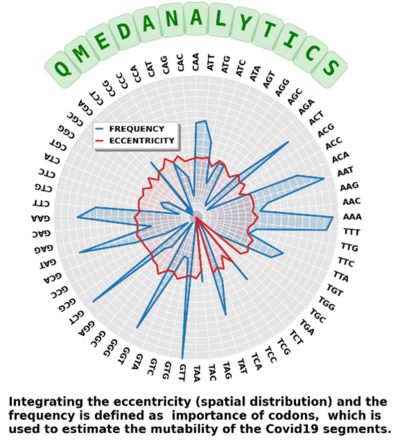
Researchers at the university of Waterloo have developed an AI software based on a text-mining method that estimates the mutability of genomic segments of a virus directly from a reference (ancestral) whole genome sequence. The method relies on calculating the importance of genomic segments based on their spatial distribution and frequency over the whole genome. The AI software was validated by performing a large-scale analysis of the viral mutations in nearly 80,000 publicly available SARS-CoV-2 predecessor whole genome sequences. The segments identified by the software as “important” were strongly correlated with the conserved sequences that had been identified through standard/conventional mutational analysis which deploy pairwise alignments.
Advantages
- Generic method (applicable to all pathogens)
- Works with the first identified genome (no need to wait for gathering genomes from other infected individuals to analyze mutations). This will significantly reduce the time it takes to develop targeted therapeutics for a novel pathogen.
- Identifies “important” segments across several length scales including at the level of codons, genes, or gene coding regions.
Potential applications
- Rapid development therapeutics/vaccines for novel pathogens e.g., timely response to highly infectious novel pathogens that have the potential to cause widespread epidemics or global pandemics.
- Identify potential candidates for stable siRNA-based targeted drugs inhibiting the production of viral proteins.
- Potential identification of novel therapeutic strategies to help overcome antimicrobial resistance.
Reference
10184
Patent status
US Patent Pending
Stage of development
Prototype built
Ongoing research
Contact
Scott Inwood
Director of Commercialization
Waterloo Commercialization Office
519-888-4567, ext. 43728
sinwood@uwaterloo.ca
uwaterloo.ca/research