Our bodies are made of trillions of cells that form tissues and organs. The genes inside the nucleus of each cell code for proteins that determine a cell’s structure and function, as well as instruct a cell when to grow, divide and die. Normally, our cells follow these instructions, but if a cell’s DNA mutates it can cause the cell to divide and grow out of control. Cancer is fundamentally a disease of uncontrolled cell growth and regulation, and all cancers ultimately are caused by mutations to the genes that regulate cell division, growth and differentiation.
Our immune system defends the body against harmful pathogens such as bacteria and viruses, but it also protects us against cancer by eliminating tumours as they form. Specialized cells of the immune system called T cells can detect cancer cells and destroy them. The immune system’s ability to find and kill cancer cells is the basis for a revolutionary kind of treatment known as cancer immunotherapy — a treatment clinicians use to strengthen the T cell’s response to cancer.
But to understand how T cells recognize cancer cells requires a short explanation of how DNA is translated into proteins.
The DNA in our genes contains information to build proteins — the molecules in our cells that carry out all functions necessary for life — but DNA does not create proteins directly. The flow of genetic information from DNA in the cell’s nucleus to the proteins that are synthesized within the cell involves two major steps called transcription and translation.
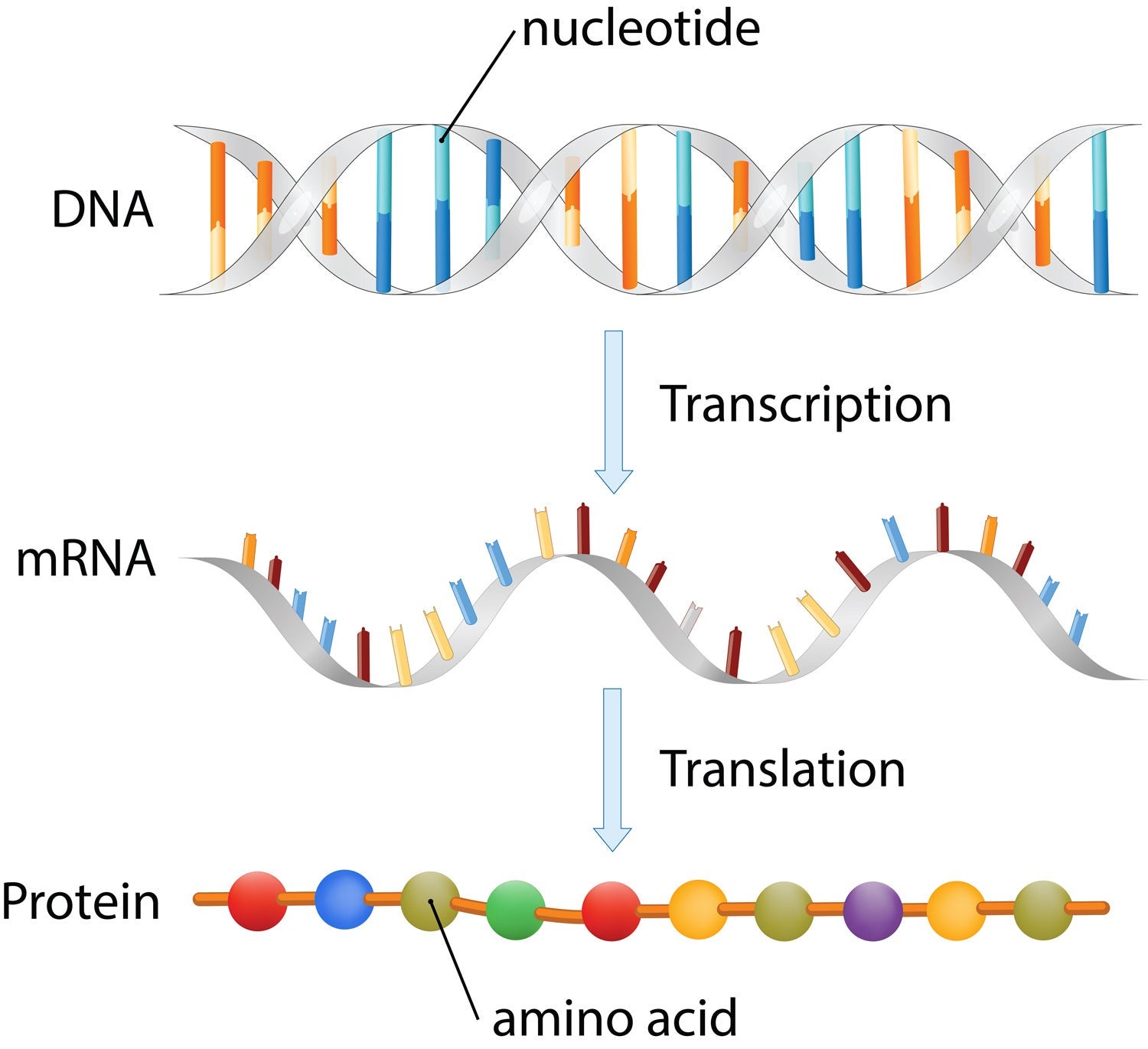
The process by which DNA is copied to mRNA is called transcription, and the process by which mRNA is used to synthesize proteins is called translation.
Transcription is the first step in a gene’s expression, the process by which information from a gene is used to synthesize a protein. During transcription, the DNA of a gene serves as a template to create messenger RNA, also known as mRNA, which is a single-stranded molecule composed of nucleotides that correspond to the genetic sequence of a gene.
The mRNA copy of a gene’s DNA sequence carries the information needed to build a protein, a large molecule made of many amino acids. During translation, the mRNA is read by cellular enzymes as a genetic code that relates the nucleotide sequence of DNA in a gene to the sequence of amino acids that form a protein molecule.
If a mutation occurs in a cell’s DNA — even if just one nucleotide is substituted for another in a gene — it affects DNA’s transcription to mRNA and the mRNA’s translation to the sequence of amino acids.
This is where the immune system’s ability to recognize non-self comes in. If the protein synthesized by a cell is changed, our immune system sees it as non-self. And using the same mechanism that T cells use to recognize pathogens in the body as foreign or non-self, T cells target and kill cancer cells that present neoantigens, small pieces of non-self protein on their surfaces.
“In cancer, when a missense mutation occurs in a cell’s DNA, a single nucleotide substitution results in a different amino acid during translation. As a result, the peptide — a fragment of the protein — that carries the mutated amino acid can be recognized by our immune system as foreign, even though it is synthesized by the cancer cell from our own body,” explains Hieu Tran, an Adjunct Assistant Professor at the Cheriton School of Computer Science and Senior Research Scientist at Bioinformatics Solutions.
“This mutated peptide is what is called a neoantigen — a new antigen that’s present only on the surface of cancer cells, but not normal cells. Our immune system can recognize neoantigens on cancer cells and kill those cancer cells, while not affecting the normal cells. We can use these same neoantigens to develop cancer vaccines to boost our immune system to eliminate a tumour.”
“When a cell becomes cancerous the human leukocyte antigen or HLA system knows about it,” adds Ming Li, a University Professor at the Cheriton School of Computer Science, who also holds the Canada Research Chair in Bioinformatics. “The HLA system cuts the mutated protein into peptides and presents those peptides on the surface of the cell. If the HLA presents a normal peptide, the T cells know that it is a self-peptide and they don’t attack it. They attack only cells with mutated peptides — the neoantigens — that are not recognized as self.”
When a tumour is found in the body, a surgeon will remove a sample for analysis. Then using a technique known as mass spectrometry, which identifies molecules based on their mass, the amino acid sequence of the neoantigens on the surface of the tumour cells can be determined, Dr. Tran said.
The trick, however, is finding the tumour-specific neoantigens — essentially a needle in a large haystack. Not surprisingly, it is a bewilderingly difficult task to do using conventional methods, but it is crucially important because neoantigens are the non-self peptides that the immune system uses to find and destroy cancer cells.
Amino acids are the building blocks of peptides and ultimately proteins. Although many amino acids have been identified in nature, just 20 amino acids make up the proteins found in the human body. By convention, amino acids are labelled using a one-letter code. For example, the amino acid alanine is labelled A, arginine is labelled R, asparagine is labelled N, and so on. A peptide’s amino acid sequence can be considered as a word of composed of these letters.
“If you are familiar with Natural Language Processing, you’ve likely seen your mobile phone guess the next word you might have typed as you compose a message. You write ‘how’ and it suggests ‘are’ and if you type ‘are’ it suggests ‘you’,” Dr. Tran said.
“We applied a similar machine-learning model to determine the amino acid sequence of neoantigens based on this one-letter amino acid code. We predict the peptide’s sequence by predicting its amino acids one at a time. If I know your immunopeptidome — the thousands of short 8 to 12 amino acid peptide antigens displayed on the cell surface — and I know that a neoantigen is different from your existing peptides by just one mutation, I can train a machine learning model using your normal peptides to predict the mutated peptides. We used a recurrent neural network — a machine learning model we call DeepNovo — to predict the amino acid sequence of neoantigens one letter at a time.”
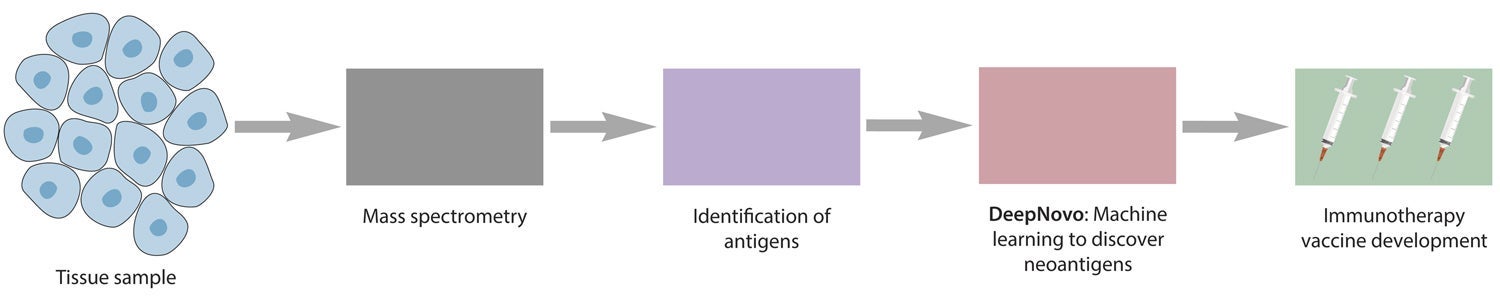
Personalized de novo peptide sequencing workflow to discover neoantigens to develop cancer vaccines (view larger figure).
{kind=link}
To do this the researchers downloaded the immunopeptidome datasets of five patients with melanoma, a type of skin cancer, which they then used to train, validate and test their machine learning model.
“Our machine-learning model expanded the predicted immunopeptidomes of those patients by 5 to 15 percent using only the data from mass spectrometry,” Dr. Tran said. “We also discovered neoantigens, including those with validated T-cell responses that had not been reported in previous studies.”
Even more impressively, the machine learning model is able to personalize the results — that is, it identifies specific neoantigens for each individual patient.
“The most exciting thing is that our approach is truly personalized — personalized to each individual patient as opposed to a group of similar patients. We used the data of each individual patient to identify his or her own neoantigens and develop a cancer vaccine specifically for that patient,” Dr. Tran said.
“Cancer immunotherapy is quickly becoming a fourth modality of cancer treatment, alongside surgery, chemotherapy and radiotherapy,” adds University Professor Li. “Every patient is different and every cancer is different, so cancer treatment shouldn’t be the same for all. Treatment should be tailored to the patient, and that’s what our personalized machine learning model allows us to do.”
Ming Li (L) is a University Professor at the Cheriton School of Computer Science and the Canada Research Chair in Bioinformatics. He is known for his fundamental contributions to Kolmogorov complexity, bioinformatics, machine learning theory, and analysis of algorithms. Hieu Tran (R) is an Adjunct Assistant Professor at the Cheriton School of Computer Science and Senior Research Scientist at Bioinformatics Solutions, a Waterloo-based company that uses machine learning to sequence and identify proteins.
To learn more about the research on which this feature is based, please see the following scientific journal publications.
Ngoc Hieu Tran, Rui Qiao, Lei Xin, Xin Chen, Baozhen Shan, Ming Li. Personalized deep learning of individual immunopeptidomes to identify neoantigens for cancer vaccines. Nature Machine Intelligence 2, 764–771 (2020).
Ngoc Hieu Tran, Rui Qiao, Lei Xin, Xin Chen, Chuyi Liu, Xianglilan Zhang, Baozhen Shan, Ali Ghodsi, Ming Li. Deep learning enables de novo peptide sequencing from data-independent-acquisition mass spectrometry. Nature Methods 16, 63–66 (2019).
Ngoc Hieu Tran, Xianglilan Zhang, Lei Xin, Baozhen Shan, Ming Li. De novo peptide sequencing by deep learning. Proceedings of the National Academy of Sciences 114 (31), 8247–8252 (2017).